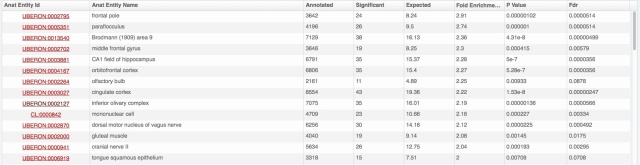
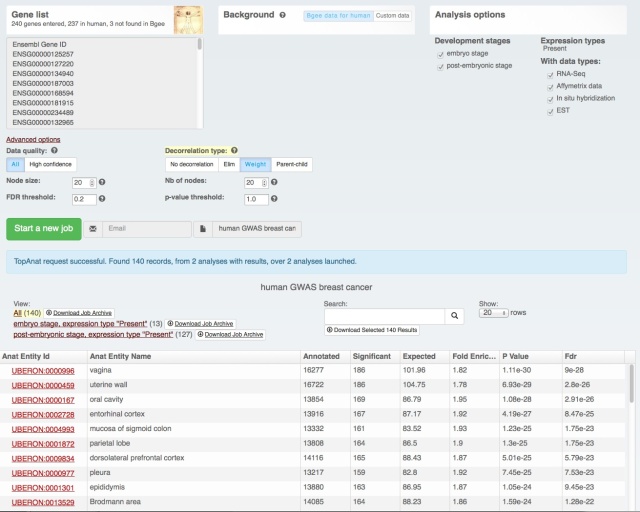
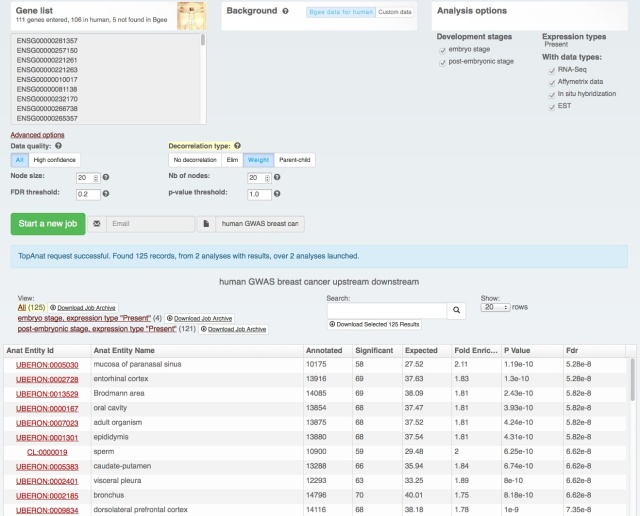
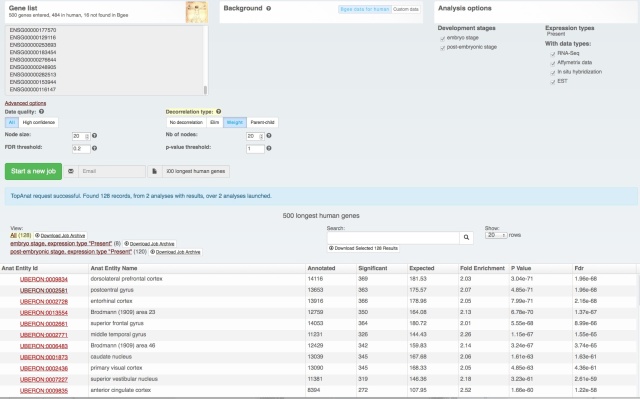
Yesterday we have released a major update of our web interface:
The download functionality remains the same, as does the data (processing for the next release is just starting with the new Ensembl release – we love Ensembl). It’s just easier to find and use (we hope! Feedback welcome at bgee@sib.swiss).
We also rolled out some improvements in TopAnat:
- clearer access to documentation;
- clearer access to example datasets;
- correction of a bug in column sorting.
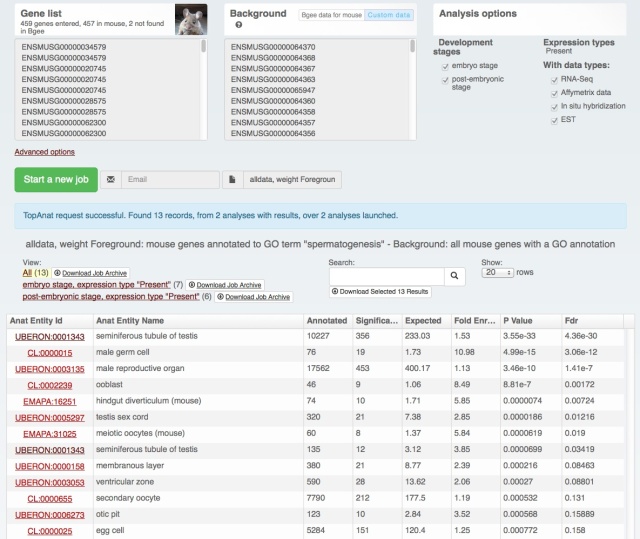
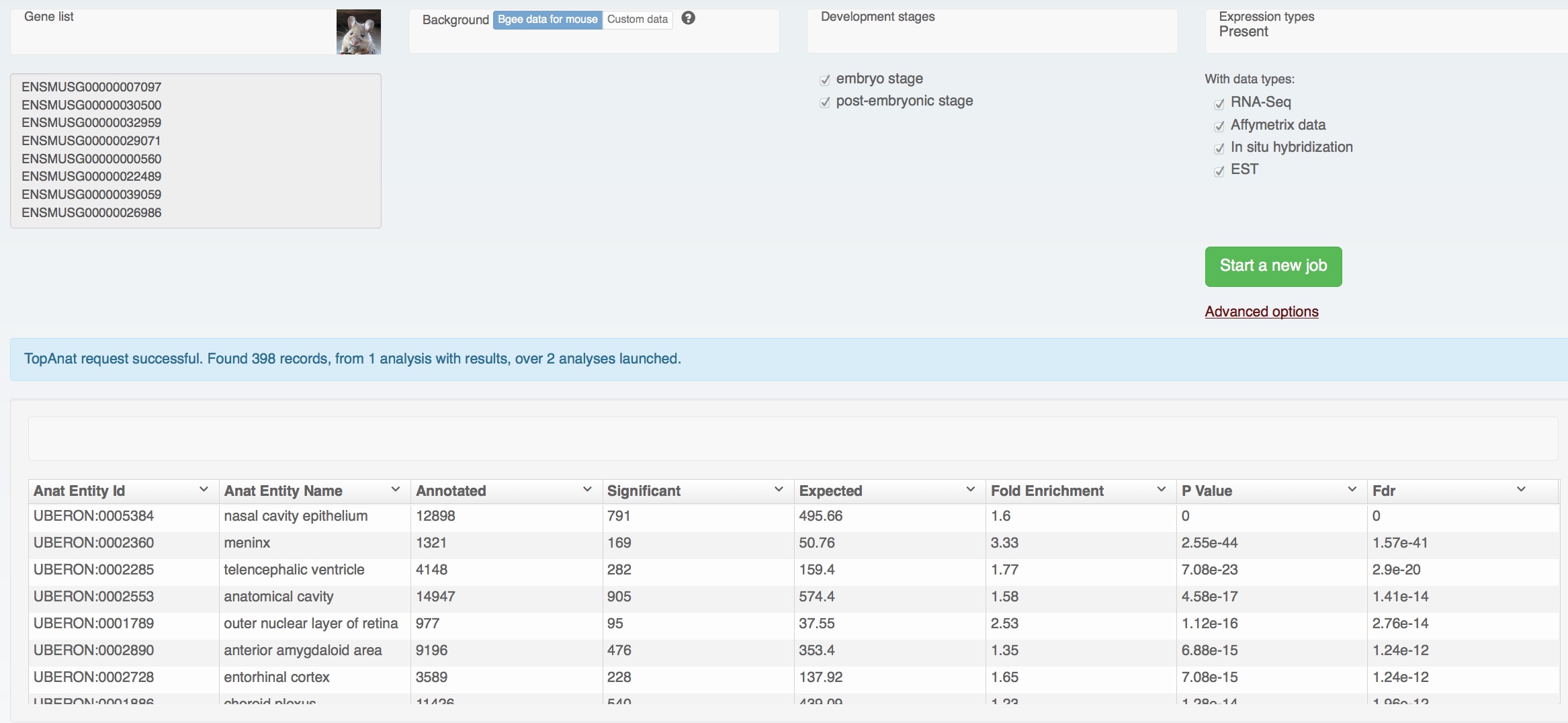
But the major functional improvement is the new gene page (click on the picture, or on “Gene search” on the Bgee homepage):
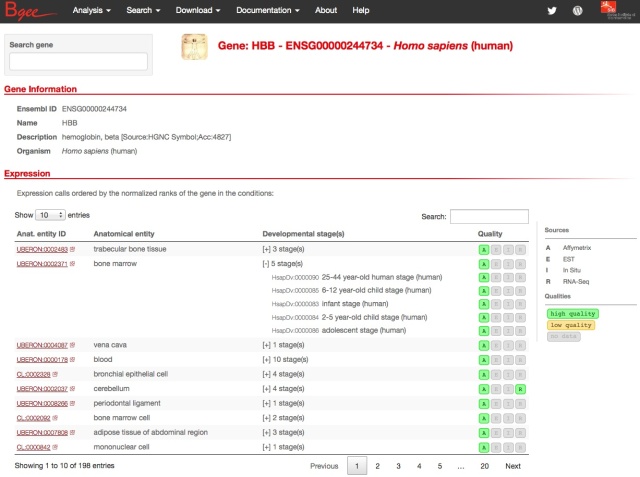
All expression information for each gene is summarized. It may look easy, but it took us quite a bit of thinking, and programming, and re-thinking, and re-programming, to find a presentation which satisfied us.
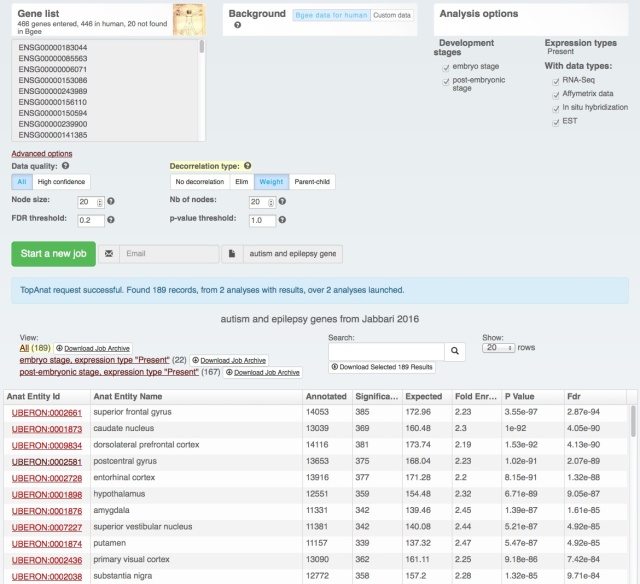
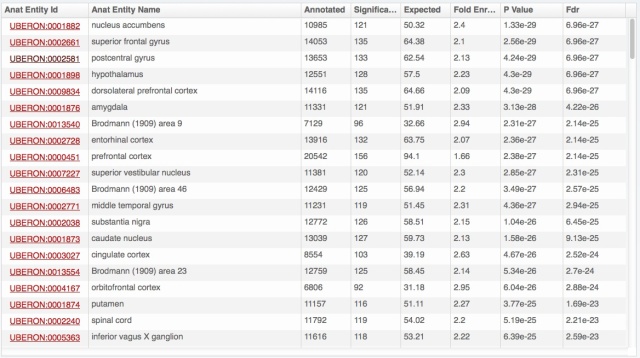
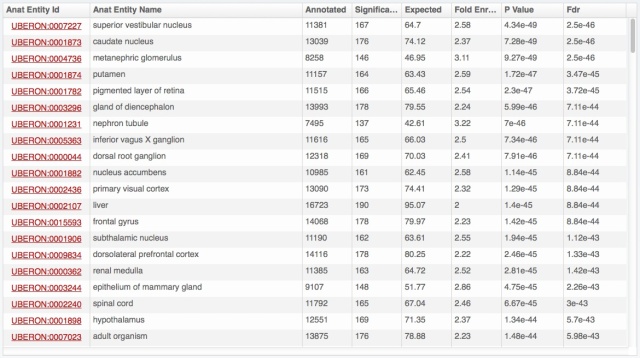
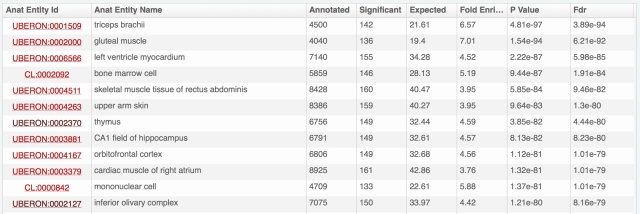
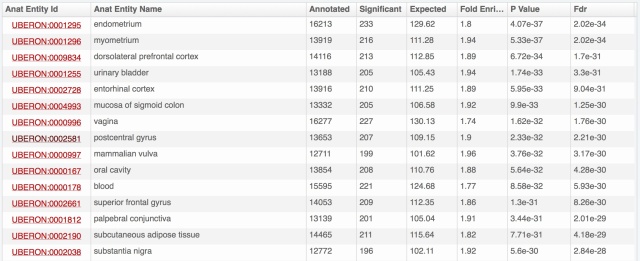
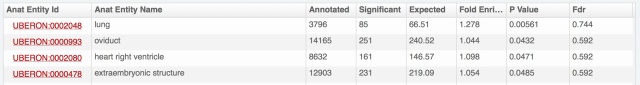
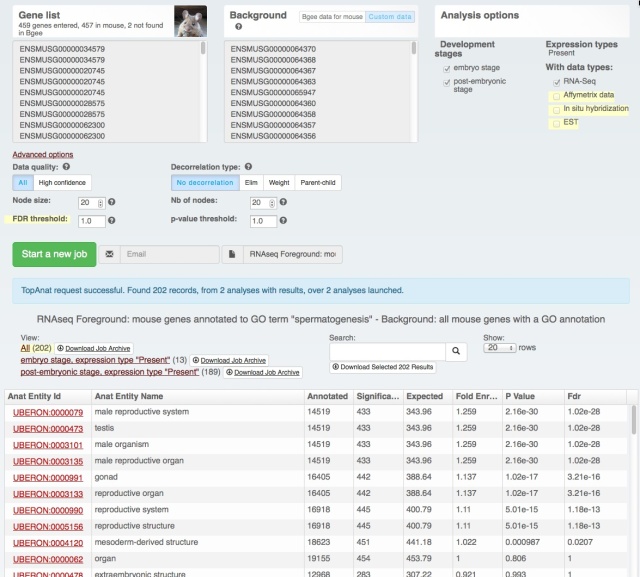
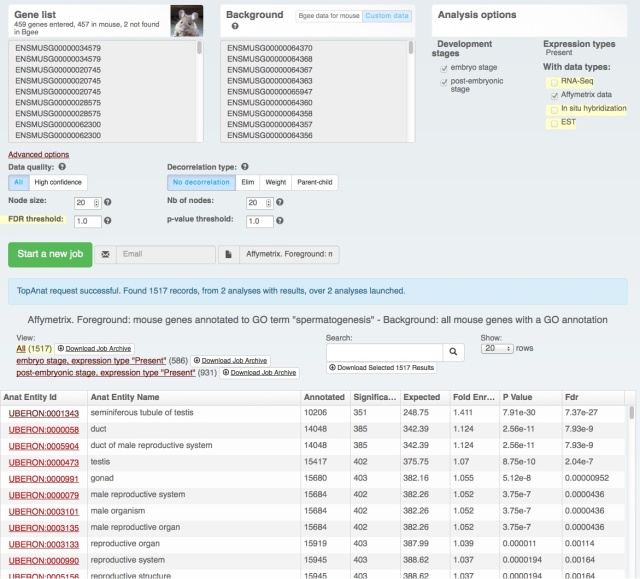
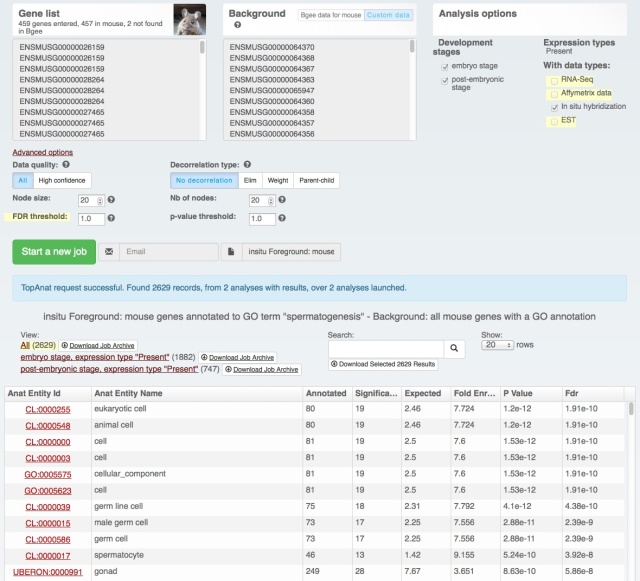
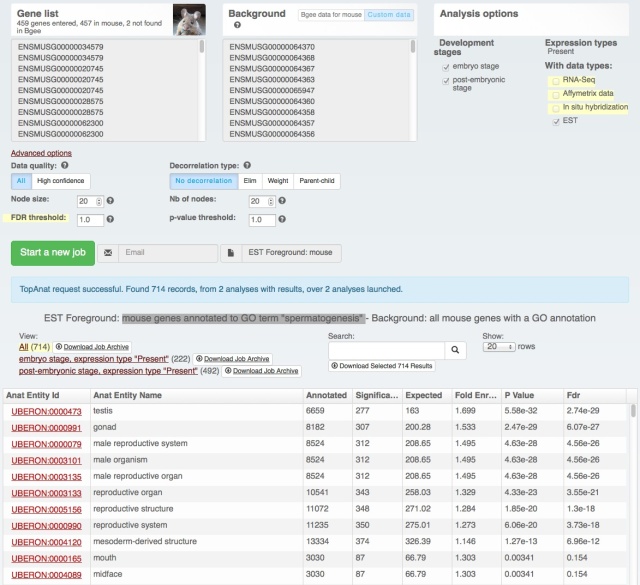
The problem is that we have really a lot of information on expression patterns for each gene, especially in major model species. Hemoglobin beta of human, presented above, is expressed in 696 combinations anatomical structure – life stage; 8108 with full propagation in the anatomical ontology. And for each of these, we may have several evidence lines, from different types of expression data. Yet we know that for this gene the most important expression is in blood cells, thus bone marrow, blood or heart should come on top. Showing the amount of data from the download files:
> grep ENSG00000244734 Homo_sapiens_expr-simple.tsv | wc
696 9249 83866
> grep ENSG00000244734 Homo_sapiens_expr-complete.tsv | wc
8108 266136 1678370
The challenge is to summarize this without hiding too much of the relevant information, but while putting the most important information forward clearly. For this, we made the following choices:
- anatomical structures are put forward by default; life stages (development and aging) are available by unfolding for each anatomical structure (as illustrated above with bone marrow).
- for each anatomical structure, we compute a normalized rank. This is rather complicated, as we need to weight for the fact that RNA-seq typically covers all genes, and in situ hybridization only a few, with microarrays and ESTs in between. We start by ranking genes inside each experiment. Briefly, the maximum rank is used as a normalizing factor. Maximum rank takes into account the number of genes called, but also how well they are differentiated: if 100 genes are called by in situ hybridization in a condition, but 99 have 1 evidence each and 1 gene has 10 evidence lines, the max rank is lower than if 100 genes each have different numbers of ESTs. For Affymetrix there is a normalization between experiments for a same condition. For in situ hybridization, all evidence for a given condition is put together as one “pseudo-experiment”, and the number of lines of evidence for a gene is used for ranking. Then each gene gets a mean rank per data type and per condition, and these are normalized. And these are then averaged for a condition, weighting again by max ranks. We forgive you if you didn’t completely follow. The important things are: do the most relevant conditions come on top? (They seem to.) Is the data behind it available? (Yes, see the download pages.)
- call quality is not taken into account, as we did not find that it added anything to the ranking; this might lead us to reconsider our evaluation of call quality in the future, but that’s another story.
- expression data is presented simply in the form of little vignettes indicating that a type of data was used to call a condition.
This is the first version of this gene page, and obviously more will need to be added to it, such as links to the data, homology links, links to other databases, and more elaborate search features.
More details on the algorithm and comparisons to other databases on Marc’s blog.
But we are already very proud of how well the ranking algorithm works. It was tested on several genes, such as hemoglobin, and we consistently get the most biologically relevant structures on top. Importantly, this is done without sacrificing specificity: we do not necessarily push to the top simply the more general anatomical terms, such as “vascular system”. And while the weighting scheme down weights in situ data when it is scarce and thus uninformative, good in situ data can push anatomical structures to the top of this list, as in the example of zebrafish insulin expression in the pancreas.